多くの実験科学者が計算化学もこなす現在、化合物の構造最適化くらいは誰でもできるという時代になってきました。
しかし、大変残念なことに、学会等を見ていると計算方法や解析方法が正しくないことも少なくありません。
例えば、水素結合や  相互作用などに代表される非共有結合相互作用 (Non-Covalent Interaction) です。
相互作用などに代表される非共有結合相互作用 (Non-Covalent Interaction) です。
X 線結晶構造などを見て “原子 A と 原子 B は X の距離にあるので相互作用があります” という考察は理解することができます。しかし、計算で得られた座標をもとに同じことを言っている人を見ると大変残念な気持ちになります。計算化学は、エネルギーを算出できるのだから、もっと定量的考察が求められるべきだと思います。
そこで、今回は 非共有結合相互作用 (Non-Covalent Interaction) に関しての論文と計算方法を少しだけ紹介したいと思います。
定量的な議論
計算化学では、相互作用があるか無いかという定性的な考察でなく、X kcal/mol の相互作用があるという定量的な考察をする方が良いと思います。
例えば、NBO 解析などで相互作用の強さを X kcal/mol と示したり、AO を視覚化してどの程度軌道がオーバーラップしているかを示したり、complex を形成することにより X kcal/mol の安定化が起こるなどを示さなければいけないと思います。
(参考:NBO解析のinputファイル作成方法
NBO解析の結果の見方)
非共有結合相互作用 (Non-Covalent Interaction) の視覚化
NBO 計算の結果を視覚化するというのは、非常に良い手法なのですが、AO を一つずつ選択していかなくてはいけません。低分子なら大した手間ではありませんが、タンパク質とリガンドの相互作用など広範囲に及ぶものでは、非常に大変です。ノートパソコンだと、きっとフリーズすると思います。
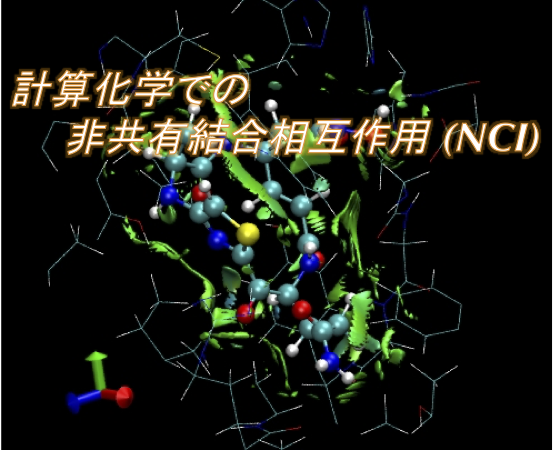
そこで、NCI の視覚化には、例えば NCIPLOT のようなものを使うことをお勧めします。
NCIPLOT は 2011 年に Julia Contreras-Garcia らにより発表されたソフトです。下図のように相互作用を視覚化することができます。(画像は、こちらのページより転載。)

インストール方法はすごく簡単であり、こちらのページからダウンロードし、お手持ちのパソコン上で解凍します。その後、src ディレクトリ内で make コマンドを実行し、パスを通せばインストール完了です。
input ファイルには、拡張子が .nci となっているものを用います。下記のコマンドで計算を実行します。
nciplot < inputfile.nci > outputfile.nco
計算が完了すると cube ファイルや .vmd ファイルが生成します。これらのファイルを vmd というソフトで可視化します。
まず、vmd で 二つの cube ファイルを読み込みます。次に、File > load visualization state で vmd ファイルを読み込みます。
続いて、Graphics > Representation を選択し、Create Rep というボタンを押すと、NCI が可視化できます。
非常に便利なソフトなのですが、相互作用を実際に計算するため、パソコンの性能や分子の大きさによって計算時間が長くなることもあります。私の自作 PC (Core i7 7700, 3.6 GHz, メモリ 32 GB) と MacBooc Pro (Core i5-5257U, 2.9 GHz, メモリ 16GB) で比べても 5 倍程度計算時間が違いました。MacBooc Proの方では、ファンが猛烈な勢いで回っていたので、途中で止めようかとも思ってしまいました。。。
GPU による高速化
先日、JCC を眺めていたら GPU を用いて NCI 計算を加速するという論文がありました。最近管理人は、自作 PC を組んだりして GPU がマイブームだったので、目を引かれました。
“GPU accelerated implementation of NCI calculations using promolecular density”
Gaëtan Rubez, Jean-Matthieu Etancelin, Xavier Vigouroux, Michael Krajecki, Jean-Charles Boisson and Eric Hénon
J. Comput. Chem. in press. DOI: 10.1002/jcc.24786
NCIPLOT は input ファイルの情報に基づき相互作用がある場所を計算しています。それほど時間のかかる計算ではありませんが、GPU で加速できたら便利ですね。(sample ファイルのフェノール二量体の相互作用を計算するので、自作 PC だと 50 秒程度でした。閾値の設定にもよりますが、タンパク質ーリガンド間の相互作用となるともっとかかりそうです。)
NCIPLOT はグリッドベースの計算であるため、グリッドサイズやインクリメントの設定が重要となります。これまでは、0.1 bohr (0.053 Å) がデフォルト値として用いられてきましたが、0.04 bohr (0.021 Å) の方が良いという報告もあります。今回は 0.025、0.05、0.1、0.2 Åで検討が行われました。
今回の論文の検討結果では、0.025 Å サイズだと リガンド96原子、タンパク 1341 原子くらいが上限っぽい感じだそうです。グリッドの数と構成原子数が大きくなっていくと GPU のメモリーの大きさが重要になってくるそうです。
今回は Pascal 世代の GPU は試されていないのでメモリの上限は 6GB らしいのですが、P100 などになれば 16GB なので、さらに巨大な系であるタンパクータンパクの相互作用の計算も行えるようになるかもしれませんね。
GPU を利用したことにより計算速度は 39 倍速くなり、電力消費も大幅に減っています。
参考文献
- Julia Contreras-Garcia Group HP
- “GPU accelerated implementation of NCI calculations using promolecular density”
Gaëtan Rubez, Jean-Matthieu Etancelin, Xavier Vigouroux, Michael Krajecki, Jean-Charles Boisson and Eric Hénon
J. Comput. Chem. in press. DOI: 10.1002/jcc.24786