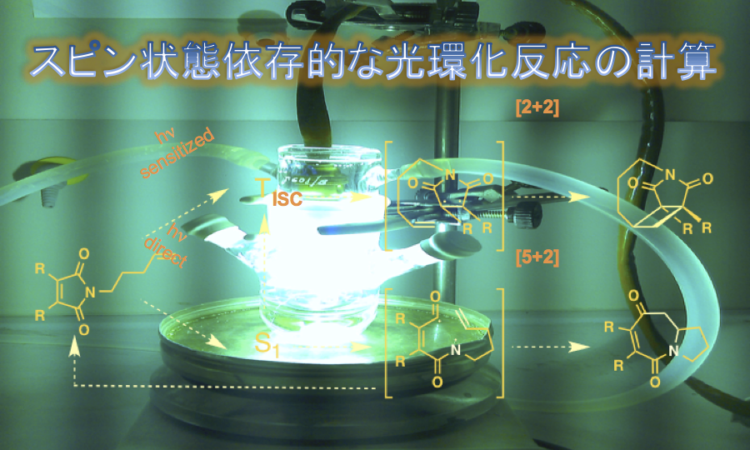
スピン状態依存的に異なる反応性を示す光環化反応について計算化学を用いた解析が行われました。JCC に論文が出ていましたので、簡単に紹介します。
“Theoretical Studies of Spin State-Specific [2 + 2] and [5 + 2] Photocycloaddition Reactions of N-(1-penten-5- yl) maleimide”
Xiang-Yang Liu, Pin Xiao, Wei-Hai Fang, and Ganglong Cui, J. Comput. Chem. 2017, in press, DOI: 10.1002/jcc.24897
概要
N-alkenyl maleimide は、[2 + 2] および [5 + 2] 光環化付加に対してスピン状態に特異的な化学選択性を示すことが見出されている。しかし、反応メカニズムは依然として不明である。この研究では、高レベルの計算手法(DFT、CASSCF、およびCASPT2)を用いて、 状態および
状態および  状態の N-alkenyl maleimide の [2 + 2] および [5 + 2] 光環化付加反応経路を探索した。 状態では [5-2] 光環化付加反応には活性化障壁がなく、圧倒的に支配的であることが分かった。[2 + 2] 光環化付加反応はその大きな障壁のために、 状態では重要ではない。反対に、 状態では [2 + 2] 光環化付加反応は、[5 + 2] 光環化付加反応よりもずっと有利である。反応機構としては、 [5 + 2] および [2 + 2] 光環化付加反応の両方をもに stepwise に進行する。 [5 + 2] 光環化付加反応において、ethenyl 部分の二級炭素が最初に maleimide 部分の窒素原子と反応し、 中間体を形成する。その後、内部転換により
状態の N-alkenyl maleimide の [2 + 2] および [5 + 2] 光環化付加反応経路を探索した。 状態では [5-2] 光環化付加反応には活性化障壁がなく、圧倒的に支配的であることが分かった。[2 + 2] 光環化付加反応はその大きな障壁のために、 状態では重要ではない。反対に、 状態では [2 + 2] 光環化付加反応は、[5 + 2] 光環化付加反応よりもずっと有利である。反応機構としては、 [5 + 2] および [2 + 2] 光環化付加反応の両方をもに stepwise に進行する。 [5 + 2] 光環化付加反応において、ethenyl 部分の二級炭素が最初に maleimide 部分の窒素原子と反応し、 中間体を形成する。その後、内部転換により  状態になる。 [2 + 2] 光環化付加反応では、ethenyl 部分の末端炭素原子は最初にmaleimide の炭素原子を攻撃し、続いて交換交叉( –> ) により 状態の中間体を与える。今回の計算結果は実験結果を合理化するだけでなく、反応機構に関して新たな知見を与えた。
状態になる。 [2 + 2] 光環化付加反応では、ethenyl 部分の末端炭素原子は最初にmaleimide の炭素原子を攻撃し、続いて交換交叉( –> ) により 状態の中間体を与える。今回の計算結果は実験結果を合理化するだけでなく、反応機構に関して新たな知見を与えた。
計算レベル
中間体の構造最適化に関しては、 と のは UB3LYP-D が用いられ、 と の遷移状態の構造最適化には CASSCF (10 electrons in 8 orbitals) が、 の遷移状態構造最適化には UB3LYP-D3 が用いられました。また、一点計算にはCASPT2 が用いられました。PCM で アセトニトリルの溶媒効果が見積もられています。
基底関数は 6-31G* が用いられています。UB3LYP-D を用いた計算には gaussian09 が、CASSCF と CASPT2 の計算には MOLCAS8.0 が用いられました。
感想
もともとの反応が報告された論文は参考文献 1。
個人的に、スピン状態が変化する反応やラジカル反応の計算に非常に興味を持っていますし、Crossing-point optimizations ってどのようにするんだろうって疑問でした。
今回は、”Penalty-function-based two-state crossing-point optimization” という方法を用いたようです。数式的には以下のように表されるようです。詳しくは、参考文献2-4。
![f(R) = \frac{E_I+E_J}{2} + c_1c_2^2\rm{ln}[1 + {(\frac{E_J-E_I}{c_2})^2]](http://computational-chemistry.com/top/wp-content/ql-cache/quicklatex.com-ce5722f42d60db46aef2e60a82df44d6_l3.png "Rendered by QuickLaTeX.com")
個人的に知る限りでは、Cross-point の計算は非常に時間がかかると聞いたことがあるのですが、この方法ではどの程度の計算コストなのか気になるところです。

状態では、もっとも低い遷移状態でも 37 kcal/mol 程度のエネルギーが必要であり、光励起なしでは反応が進行しないことがわかります。
実験結果と計算結果がきちんと相関しており、これほどきれいに結果が出るもんなんだと驚きました。
参考文献
- “Reaction Control in Synthetic Organic Photochemistry: Switching between [5+2] and [2+2] Modes of Cycloaddition”
C. Roscini, K. Cubbage, M. Berry, A. Orr-Ewing, K. Booker-Milburn,
Angew. Chem. Int. Ed. 2009, 48, 8716-8720. DOI: 10.1002/anie.200904059 - “The Photoisomerization Mechanism of Azobenzene: A Semiclassical Simulation of Nonadiabatic Dynamics” C. Ciminelli, G. Granucci, M. Persico, Chem. Eur. J. 2004, 10, 2327-2341. DOI: 10.1002/chem.200305415
- “DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations”
J. Kastner, J. Carr, T. Keal, W. Thiel, A. Wander, P. Sherwood, J. Phys.
Chem. A 2009, 113, 11856-11865. DOI: 10.1021/jp9028968 - “ChemShell—a modular software package for QM/MM simulations”
S. Metz, J. K€astner, A. A. Sokol, T. W. Keal, P. Sherwood, WIREs Comput. Mol. Sci., 2014, 4, 101-110. DOI: 10.1002/wcms.1163
関連する記事
- Threadripper の Gaussian16 ベンチマーク
- Threadripper 自作 PC 組み立て編【AMD】
- プロパギルアミンと二酸化炭素を基質とした銀触媒による 5/6 員環形成反応の選択性に関する計算
- 歪曲・相互作用モデル distorsion/interaction model
- 密度汎関数法の基礎 (KS物理専門書)
- 計算化学者なら知っておいた方が良いパソコンの知識
- Fedo25 に CUDA ToolKit 8.0 をインストールしてみた
- スパコンランキング発表!日本はGREEN500上位独占!【2017年6月】
- 【AMD_Naples】正式名称はEPYCに決定!クロック数は2.8 GHz【基本スペックなど】
- 過酸化水素の O–O 結合のホモリシスの計算!