
有機触媒を用いた nitromethane の cinnamaldehyde へのマイケル付加の理論解析の論文が J. Phys. Chem. A に出ていました。今回の論文では溶媒効果や触媒再生機構を計算機上でうまく再現できていると思います。特に、DCM と水を用いた時の差や、水を添加した時に律速段階が変化したりエナンチオ選択性や収率が落ちることを合理的に説明しています。
“Reaction Mechanism of Organocatalytic Michael Addition of Nitromethane to Cinnamaldehyde: A Case Study on Catalyst Regeneration and Solvent Effects”
Katarzyna Świderek, Alexander R. Nödling, Yu-Hsuan Tsai, Louis Y. P. Luk & Vicent Moliner J. Phys. Chem. A in press. DOI:10.1021/acs.jpca.7b11803
概要
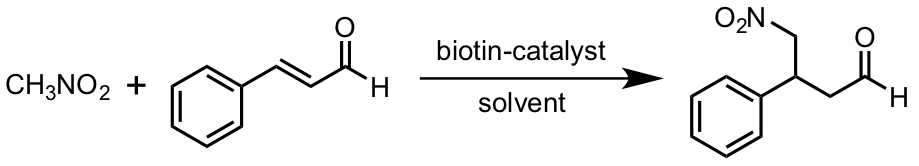
本論文では、実験と計算の両面から nitromethane の cinnamaldehyde へのマイケル付加反応を触媒存在下 & 非存在下で解析した。有機触媒としてはビオチン化された二級アミンが用いられている。計算には M06-2X 汎関数が用いられ、2つの異なる溶媒:ジクロロメタン(DCM)および水の効果を見積もるために分極可能な連続体モデル (SMD) が用いられた。一般的な仮定とは対照的に、生成物に近い構造のイミニウム中間体は、今回の計算で用いられたいずれの溶媒を用いた場合の計算結果にも存在しなかった。代わりに、エナミン中間体中の C1-C2 二重結合を水和することにより生成物を形成し、触媒を再生するための鍵となる四面体中間体が直接得られる。DCM 中では、エナミンの水和反応は協奏的であり律速段階であるが、水中ではこの反応は律速段階ではない 2 段階の反応になる。さらに解析を進めたところ、溶媒としての水の使用が他の化学的ステップ、特にイミニウム中間体と求核剤との間の C–C 結合形成の重要な段階のためのエネルギー障壁をも上昇させることを明らかにした。これにより反応収率およびエナンチオ選択性の両方が低下し、このことは実験的にも示された。これらの知見は、水が添加剤として使用されるときに水がしばしば有機触媒を増強するが、溶媒として使用されるときに反応の進行を妨げる理由についての論理的説明となると考えられる。
計算手法
M06-2X/6-31+G(d,p) で計算しています。溶媒効果の見積もりは Truhlar らによって開発された solute electron density model (SMD, 参考文献 1) を用いています。全ての計算は Gaussian 09, version A. を用いて行われました。
Pyrrolidine 類縁体の有機触媒では、しばしばかさ高い置換基や水素結合を形成する置換基が用いられています。これらの置換基は水和の step に影響していると考えられていますが、今回の論文ではその影響を取り除くため、Jørgensen−Hayashi や MacMillan 触媒などの一般的なシステムは使わずに 3-aminoproline 誘導体とビオチンの複合体を使用したそうです。また、ビオチンをつけ他のは、水への溶解性の改善の狙いもあったそうです。
内容
有機触媒というと一時のブームでかなり研究された印象を持っていたのですが、今回の論文のイントロ部分によれば、生成物のリリースのメカニズムはわかっていないそうです?!これまでにも計算化学を用いた有機触媒反応の解析は行われてきたそうですが、気相中の計算であり溶媒効果の見積もりは行われていなかったそうです(参考文献 2, 3 など)。
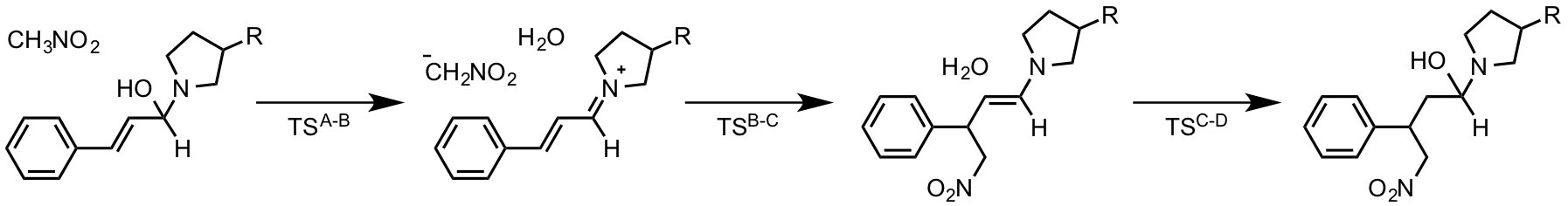
生成物に近い構造の iminium 中間体が見られなく、直接水和が起こるというのが今回の論文のポイントのような気がしますが、
Notably, the commonly assumed product-derived iminium intermediate was not observed.
の意味がはっきりしません。addredundant で TS 様の構造が見えなかったのか、計算していないだけなのか、それともエネルギーの高い経路だったのかが不明瞭です。
あと今回の論文で気になったのは計算レベルです。一段階の反応が二段階になってしまう時、溶媒によって反応機構が変わる時、これまでの報告例と反応機構が変わる場合などは、いくつかの汎関数を試した方が良いと個人的には思います。
特に M06-2X は弱い相互作用を強く見積もりすぎる傾向があるので注意した方が良いと思います。(M06-2X も solute electron density model (SMD) も Truhlar のグループによって開発されたものですが、今回の論文の著者は Truhlar 信者なのでしょうか?)
 を見ると、虚振動が -83.78
を見ると、虚振動が -83.78  と比較的小さな値です。おそらく汎関数を変えれば水を使った場合でも
と比較的小さな値です。おそらく汎関数を変えれば水を使った場合でも  と は一つの TS になるのでは?と思ってしまいます(実際に計算してみないとわかりませんが。。。)M06-2X では水の酸素原子と二重結合の
と は一つの TS になるのでは?と思ってしまいます(実際に計算してみないとわかりませんが。。。)M06-2X では水の酸素原子と二重結合の  軌道の相互作用を強く見積もりすぎな気がしてしまっています。管理人の気のせいかもしれませんが。。。
軌道の相互作用を強く見積もりすぎな気がしてしまっています。管理人の気のせいかもしれませんが。。。
ちなみに、この rate-limiting step の活性化エネルギーですが 30 kcal/mol 程度あります。反応温度は 25℃ です。30 kcal/mol でも問題なく室温条件下で反応が進行すると捉えるか、M06-2X が適切な計算レベルでないと捉えるか微妙なところだと思います。触媒非存在下ではもっとも活性化エネルギーが高いステップで 50 kcal/mol 程度あるので、こちらは反応が進行しないという結論でも良いと思います。いずれにしても、他の汎関数も検討した方が良いような気がします。
Frontier Orbital Analysis も行い、今回用いた二級アミンと基質がイミニウム中間体 (INT B) を形成することにより HOMO-LUMO ギャップが小さくなり、活性化エネルギーが下がるということが書かれていました。
また、water configuration も気になりました。水中の反応では 水分子が基質の周囲に無数に存在しているはずなので、水分子が 2 分子、3 分子関与する場合も検討すべきではないかと思いました。
あと全く関係ありませんが、論文 2 ページ目左にある trail-and-error は trial-and- error の間違いでしょうか?
記事中に間違い等ある場合は、コメント欄、twitter またはメールにてお知らせいただけると幸いです。
参考文献
- “Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions” Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378−6396. DOI: 10.1021/jp810292n
- “Computational Approach to Diarylprolinol-Silyl Ethers in Aminocatalysis”
Halskov, K. S.; Donslund, B. S.; Paz, B. M.; Jørgensen, K. A. Acc. Chem. Res. 2016, 49, 974−986. - “Computational Insights into the Central Role of Nonbonding Interactions in Modern Covalent Organocatalysis”
Walden, D. M.; Ogba, O. M.; Johnston, R. C.; Cheong, P. H. Acc. Chem. Res. 2016, 49, 1279−1291.
関連する記事
- 正宗・Bergman 環化の計算化学【エンジイン】
- ニューラルネットワークを利用した粗視化シミュレーション コンフォメーション探索
- M06 / M06-2X
- revM06-L が発表されました。
- ドナルド・トゥルーラーDonald G. Truhlar
- Threadripper 並列化効率改善?【gaussian16】
- Threadripper Gaussian16 ベンチマーク
- IRC 計算がうまくいかない時
- スピン状態依存的な光環化反応の計算
- 【Gaussian 16】デスクトップ PC で並列計算する際の注意点【Hyperthreading】
- スパコンランキング発表!日本はGREEN500上位独占!【2017年6月】
- 自作 PC を作ってみた!【OS 編】