GRRM17 の AFIR は、非常に強力な化学反応経路の探索手法です。
GRRM は非常に便利なプログラムではあるのですが、出力ファイルをそのままでは GaussView では開くことができないという少し面倒な点があります。
前回の記事では、クリップボード上の分子を可視化するプログラムを紹介しましたが、1 分子ずつしか可視化できません!
そのため、大量の構造を含んでいる出力ファイルの処理には向いていません。
そこで今回、大量の座標情報を効率的に処理することを目的として、簡単なプログラムを書きました。
GRRM の出力ファイルの一つであり EQ_list.log を GaussView で瞬時に可視化するプログラムを紹介します。
関連記事:GRRM の振動計算の結果を Gauss View で可視化する!
実行結果
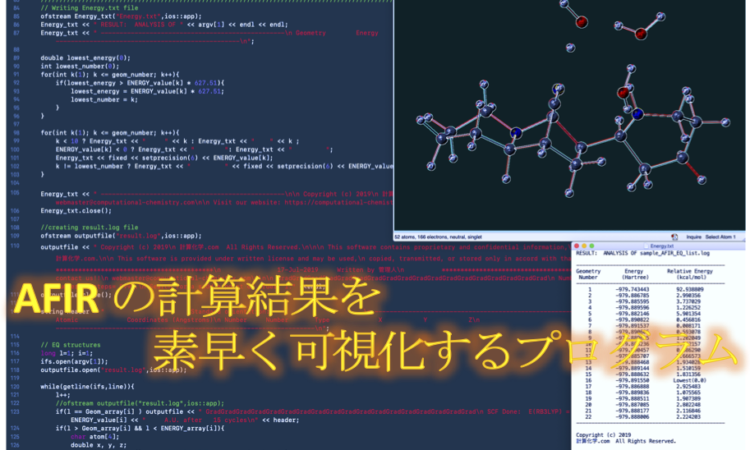
まずは、実際にプログラムを実行した時の様子をご覧ください。
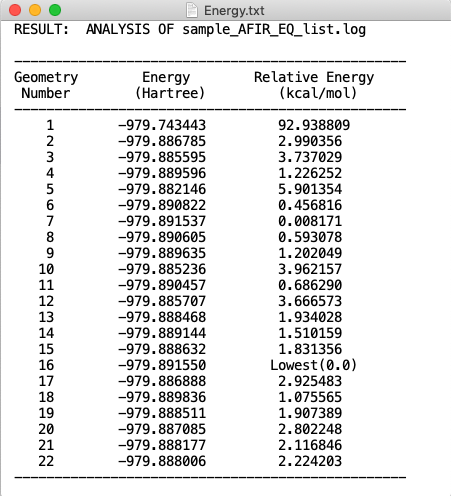
今回、使用した sample_EQ_list.log には 22 個の分子情報が入っています。これをプログラムで処理すると result.log と Energy.txt という二つのファイルが生成します。
result.log は 22 個の分子を GaussView 上で連続表示するファイルです。エネルギー情報を入っているので、GaussView の Results>Optimization から表示可能です。
また、Energy.txt にはそれぞれの分子の絶対エネルギー値(a.u.)と最安定構造を基準とした相対エネルギー値(kcal/mol)が含まれています。
プログラム内で GaussView と TextEditor の path を設定しておくと、プログラム実行後に自動でファイルを開いてくれます。
(注)result.log と Energy.txt 内の分子の番号は 1 から始まっていますが、EQ_list.log 内の分子番号は 0 から始まっています。分子番号が 1 ずれていますので、ご注意ください。
環境設定
管理人は、MacBook Pro (15-inch, 2017) を使用しています。
今回のプログラムは C++ で書きました。使用前に
g++ EQ_list.cpp
でコンパイルしてください。実際使用する際は、
./a.out sample_EQ_list.log
と入力して実行してください。
プログラムの実行に当たってエラーなど出ましたら、コメント欄で質問してください。そうすれば他の人とも情報共有できます。
実際のコード
ダウンロードしたい方は、こちらからどうぞ。

このプログラムは、TS_list.log や PT_list.log のファイルも同様に処理することができます。ファイル中の構造が、9999 個を越えるとエラーで止まりますので、この点のみご注意ください。
// written by 計算化学.com 管理人 on 17/7/2019
//
// This program reads GRRM EQ_list.log file and creats 2 files as following;
// (i)result.log (ii)Energy.txt
//
// <How to use?>
// ./a.out <filename.log>
//
// Feel free to email webmaster@computational-chemistry.com
#include <iostream>
#include <string>
#include <fstream>
#include <iomanip>
#include <ctime>
using namespace std;
int genso(string atom); //prototype declaration
// Main Function
int main (int argc, char *argv[]){
// measuring cpu time
clock_t start;
double duration;
start = clock();
// Input Error
if(argc == 1){
cout << "\n *************************\n ERROR -- no argumanet --\n ./a.out [file_name]\n *************************\n\n Copyright (c) 2019\n 計算化学.com All Rights Reserved.\n" << endl;
exit(1);
}
// Reading file name
ifstream ifs(argv[1]);
string line;
// searching start lines of each structures
int geom_number(0), Int_number(0) ,number_of_atoms(0);
int *Geom_array;
Geom_array = new int[9999];
long *ENERGY_array;
ENERGY_array = new long[9999];
double *ENERGY_value;
ENERGY_value = new double[9999];
for(int i(0); i<9999; i++){
Geom_array[i]=0;
ENERGY_array[i]=0;
ENERGY_value[i]=0;
}
long i(1);
while (getline(ifs,line)){
i++;
std::string::size_type Geom = line.find("# Geometry of");
std::string::size_type ENG = line.find("Energy =");
if (Geom != std::string::npos ){
geom_number++;
Geom_array[geom_number] = i;
}if (ENG != std::string::npos ){
ENERGY_array[geom_number] = i;
char a[10], b[2];
double energy;
sscanf( line.data(), "%s %s %lf\n", a, b, &energy );
ENERGY_value[geom_number] = energy;
}
}
ifs.close();
Geom_array[0] = Geom_array[1];
ENERGY_value[0] = ENERGY_value[1];
char filename_without_com[50];
int file_length (0);
for(int i (0); argv[1][i] !='\0'; ++i) file_length +=1;
for(int i = 0; i <= file_length-4; i++) filename_without_com[i] = argv[1][i];
filename_without_com[file_length-4] = '\0';
////////////////////////////////////////////////////////////////////////////////////////////////////////////
// Writing Energy.txt file
ofstream Energy_txt("Energy.txt",ios::app);
Energy_txt << " RESULT: ANALYSIS OF " << argv[1] << endl << endl;
Energy_txt << " -------------------------------------------------\n Geometry Energy Relative Energy \n Number (Hartree) (kcal/mol) \n -------------------------------------------------\n";
double lowest_energy(0);
int lowest_number(0);
for(int k(1); k <= geom_number; k++){
if(lowest_energy > ENERGY_value[k] * 627.51){
lowest_energy = ENERGY_value[k] * 627.51;
lowest_number = k;
}
}
for(int k(1); k <= geom_number; k++){
k < 10 ? Energy_txt << " " << k : Energy_txt << " " << k ;
ENERGY_value[k] < 0 ? Energy_txt << " ": Energy_txt << " ";
Energy_txt << fixed << setprecision(6) << ENERGY_value[k];
k != lowest_number ? Energy_txt << " " << fixed << setprecision(6) << ENERGY_value[k] * 627.51 - lowest_energy << endl: Energy_txt << " Lowest(0.0)" <<endl;
}
Energy_txt << " -------------------------------------------------\n\n Copyright (c) 2019\n 計算化学.com All Rights Reserved.\n\n If you need help, please contact us!\n webmaster@computational-chemistry.com\n\n Visit our website: https://computational-chemistry.com/top/\n\n";
Energy_txt.close();
//creating result.log file
ofstream outputfile("result.log",ios::app);
outputfile << " Copyright (c) 2019\n 計算化学.com All Rights Reserved.\n\n\n This software contains proprietary and confidential information,\n including trade secrets, belonging to 計算化学.com.\n\n This software is provided under written license and may be used,\n copied, transmitted, or stored only in accord with that written license.\n\n\n ******************************************\n 17-Jul-2019 Written by 管理人\n ******************************************\n\n\n If you need help, please contact us!!\n webmaster@computational-chemistry.com\n\n\n GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad\n Number of steps in this run= 9999 maximum allowed number of steps= 9999.\n Search for a saddle point of order 1." <<endl;
outputfile.close();
string header = " GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad\n Standard orientation: \n ---------------------------------------------------------------------\n Center Atomic Atomic Coordinates (Angstroms)\n Number Number Type X Y Z\n ---------------------------------------------------------------------\n";
// EQ structures
long l=1; i=1;
ifs.open(argv[1]);
outputfile.open("result.log",ios::app);
while(getline(ifs,line)){
l++;
//ofstream outputfile("result.log",ios::app);
if(l == Geom_array[i] ) outputfile << header;
if(l > Geom_array[i] && l < ENERGY_array[i]){
char atom[4];
double x, y, z;
sscanf( line.data(), "%s %lf %lf %lf\n", atom, &x, &y ,&z);
l - Geom_array[i]<10 ? outputfile << " " << (l - Geom_array[i]) : outputfile << " " << (l - Geom_array[i]);
genso(atom)<10 ? outputfile << " " << genso(atom) : outputfile << " " << genso(atom);
outputfile << " 0 ";
x < 0 ? outputfile << fixed << setprecision(6) << x : outputfile << " " << fixed << setprecision(6) << x;
y < 0 ? outputfile << " " << fixed << setprecision(6) << y : outputfile << " " << fixed << setprecision(6) << y;
z < 0 ? outputfile << " " << fixed << setprecision(6) << z : outputfile << " " << fixed << setprecision(6) << z;
outputfile << endl;
}if(l == ENERGY_array[i]){
outputfile << " ---------------------------------------------------------------------\n GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad\n Step number " << i << " out of a maximum of 9999\n";
i++;
}
else continue;
}
ifs.close();
outputfile.close();
//last structure of EQ_list
outputfile.open("result.log",ios::app);
ifs.open(argv[1]);
l=1; i--;
while(getline(ifs,line)){
l++;
if(l == Geom_array[i] - 1) outputfile << header;
if(l >= Geom_array[i] && l < ENERGY_array[i]){
char atom[4];
double x, y, z;
sscanf( line.data(), "%s %lf %lf %lf\n", atom, &x, &y ,&z);
l - Geom_array[i]<10 ? outputfile << " " << (l - Geom_array[i]) : outputfile << " " << (l - Geom_array[i]);
genso(atom)<10 ? outputfile << " " << genso(atom) : outputfile << " " << genso(atom);
outputfile << " 0 ";
x < 0 ? outputfile << fixed << setprecision(6) << x : outputfile << " " << fixed << setprecision(6) << x;
y < 0 ? outputfile << " " << fixed << setprecision(6) << y : outputfile << " " << fixed << setprecision(6) << y;
z < 0 ? outputfile << " " << fixed << setprecision(6) << z : outputfile << " " << fixed << setprecision(6) << z;
outputfile << endl;
}if(l == ENERGY_array[i]){
outputfile << " ---------------------------------------------------------------------\n GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad\n ! Optimized Parameters ! \n GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad\n\n Normal termination of Gaussian 16 \n";
}
else continue;
}
ifs.close();
outputfile.close();
delete[] Geom_array;
delete[] ENERGY_array;
delete[] ENERGY_value;
////////////////////////////////////////////////////////////////////////////////////////////////////////
// cpu time
duration = ( clock() - start ) / (double) CLOCKS_PER_SEC;
//cout<<"cpu time : "<< duration << " seconds"<<endl;
char buff[99];
sprintf(buff,"open /Applications/gv/gview.app result.log &"); // Please re-write the path for GaussView
system(buff);
sprintf(buff," open /Applications/TextEdit.app Energy.txt &");
system(buff);
//Copy Right
cout << "\n\n Copyright (c) 2019\n 計算化学.com All Rights Reserved.\n\n\n This software contains proprietary and confidential information,\n including trade secrets, belonging to 計算化学.com.\n\n ******************************************\n 17-Jul-2019 Written by 管理人\n ******************************************\n\n\n If you need help, please contact us!\n webmaster@computational-chemistry.com\n\n Visit our website: https://computational-chemistry.com/top/\n\n";
}
//End of Main Functional
////////////////////////////////////////////////////////////////////////////////////////////////////////
// Function converting element name to atomic number
int genso (string atom){
string elements[128]={"Error","H","He","Li","Be","B","C","N","O","F","Ne","Na","Mg","Al","Si","P","S","Cl","Al","K","Ca","Sc","Ti","V","Cr","Mn","Fe","Co","Ni","Cu","Zn","Ga","Ge","As","Se","Br","Kr","Rb","Sr","Y","Zr","Nb","Mo","Tc","Ru","Rh","Pd","Ag","Cd","In","Sn","Sb","Te","I","Xe","Cs","Ba","La","Ce","Pr","Nd","Pm","Sm","Eu","Gd","Tb","Dy","Ho","Er","Tm","Yb","Lu","Hf","Ta","W","Re","Os","Ir","Pt","Au","Hg","Ti","Pb","Bi","Po","At","Rn","Fr","Ra","Ac","Th","Pa","U","Np","Pu","Am","Cm","Bk","Cf","Es","Md","Bo","Lr","Rf","Db","Sg","Bh","Hs","Mt","Ds","Rg","Cp","Uut","Uuq","Uup","Uuh","Uus","Uus","Uuo"};
int j = 1;
if(atom.substr(1,1)==" "){
for(int i(1); i<=118; i++){
if(strcmp(elements[i].c_str(),(atom.substr(0,1)).c_str())==0){
j = i;break;
}
else continue;}
}else{
for(int i(1); i<=118; i++){
if(strcmp(elements[i].c_str(),(atom.substr(0,2)).c_str())==0){
j = i;break;
}
else continue;}}
return j;
}
一時間もかからずに書けてしまう簡単なコードですが、非常に便利なので管理人は毎日のように使っています。
GRRM17 の購入
GRRM17 は、こちらのページから購入することが可能です。アカデミックであれば 1 node 10,000 円(3 年)と格安です。
管理人は、30 node 分くらい注文しようかと考えています。