
励起状態でのプロトン移動(ESPT: Excited-State Proton Transfer )は蛍光プローブや分子スイッチ、発光ダイオードなどの分子に関係していることから近年注目されている現象です。核酸塩基間でもこの反応が進行するという結果も出ています。ほとんどの ESPT 反応では、励起により proton donor 部分の酸性度が増し、acceptor 部分の塩基性が増すことによって反応が進行するそうです。
励起状態でのプロトン移動の反応機構に関しての計算が J. Phys. Chem. B に掲載されていました。簡単に紹介したいと思います。
“Excited-State Proton Transfer Mechanism of 2,6-Diazaindoles·(H O)
O)
(n = 2−4) Clusters”
Zhe Tang, Yutai Qi, Yi Wang, Panwang Zhou, Jing Tian, & Xu Fei
J. Phys. Chem. B in press. DOI: 10.1021/acs.jpcb.7b10207
概要
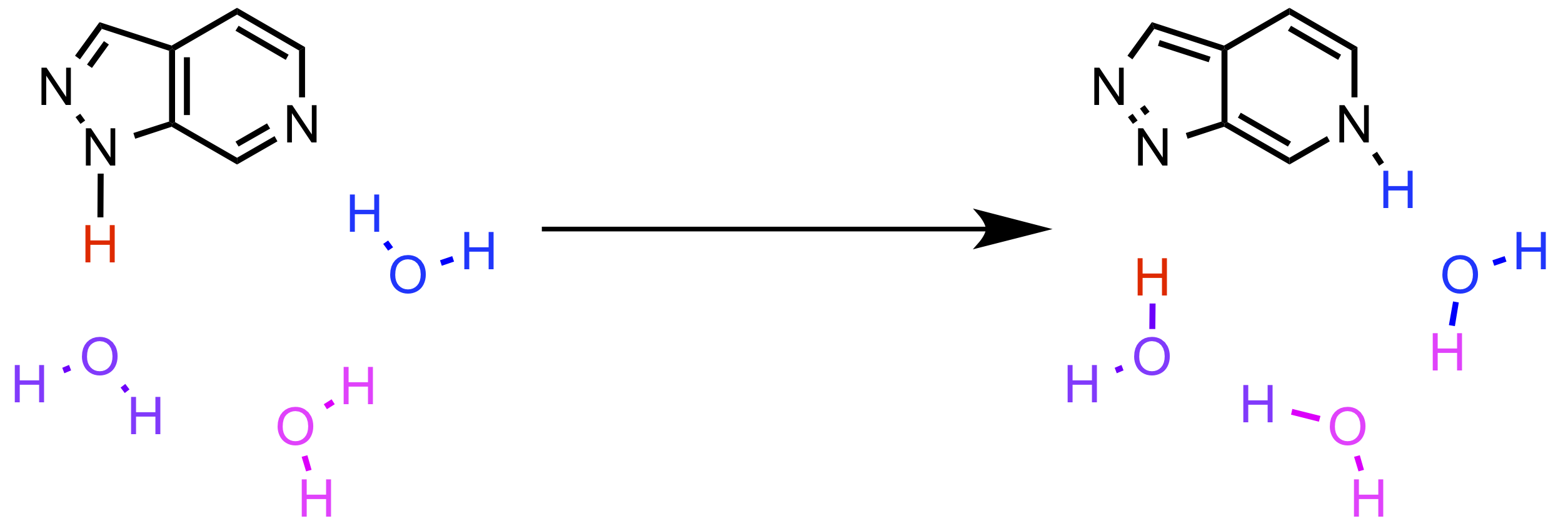
この論文では、TDDFT を用いて水溶液中での 2,6-diazaindoles(2,6-DAI)の励起状態プロトン移動(ESPT)の反応機構解析を行なっています。今回の計算結果では、2 つの水分子を有する 2,6-DAI が hydrogen bond wires を形成しないので、励起状態での 3 つのプロトンが同時に移動する反応が起こり得ないことを示しています。この知見は、以前の実験で報告されたものとは異なっています(Chungら、Chung et al. J. Am. Chem. Soc. 2017, 139, 6396−6402. DOI: 10.1021/jacs.7b01672)。 3 分子の水分子を有する 2,6-DAI は 2,6-DAI·(HO) クラスターを形成し、一方 4 分子の水分子を有する 2,6-DAI は 2,6-DAI·(HO)
クラスターを形成し、一方 4 分子の水分子を有する 2,6-DAI は 2,6-DAI·(HO) クラスターを形成します。これらのクラスターは ESPT 反応に関与しています。 2,6-DAI·(HO) と2,6-DAI·(H2O) クラスターの ESPT 機構を明らかにするために、
クラスターを形成します。これらのクラスターは ESPT 反応に関与しています。 2,6-DAI·(HO) と2,6-DAI·(H2O) クラスターの ESPT 機構を明らかにするために、 状態と
状態と  状態のポテンシャルエネルギー曲面の探索が行われました。その結果、2,6-DAI·(HO) および 2,6-DAI·(HO) クラスターの双方で、確からしいプロトン移動経路が 1 つだけ存在することが分かりました。 2,6-DAI·(H2O) および 2,6-DAI·(H2O) クラスターの遷移状態を計算することにより、ESPT 反応は一貫したメカニズムであることがわかりました。この研究は、ESPT に関与する水分子の数を調査し、生物学的分野における分子間水素結合相互作用をさらに研究するに役立つでしょう。
状態のポテンシャルエネルギー曲面の探索が行われました。その結果、2,6-DAI·(HO) および 2,6-DAI·(HO) クラスターの双方で、確からしいプロトン移動経路が 1 つだけ存在することが分かりました。 2,6-DAI·(H2O) および 2,6-DAI·(H2O) クラスターの遷移状態を計算することにより、ESPT 反応は一貫したメカニズムであることがわかりました。この研究は、ESPT に関与する水分子の数を調査し、生物学的分野における分子間水素結合相互作用をさらに研究するに役立つでしょう。
計算手法
全ての計算は、gaussian09 で行われています。, の構造最適化は B3LYP/TZVP のレベルで行われました。その後の計算は、B3LYP-D3/TZVP と水溶液中での計算ですので、IEF-PCM が使われています。(なぜ TZVP を使っているのかは謎)。
PES は、O–H の距離を徐々に変化させて作成したようです。Methods の部分に IRC に関しての記述が無いのですが、ちゃんと IRC 計算したのでしょうか?また、TS の構造で single imaginary frequency を確認したなどの記述もありませんが、どうなんでしょう?
SI にも座標やエネルギー(a.u)の情報が一切無いんですが、これで良いのでしょうか?どうやって再現しろと?
内容
今回の論文では、反応エネルギーに加えて、スペクトル計算もされています。吸収スペクトル、蛍光スペクトルの計算値が実験結果と良い一致を示していることから、今回用いた計算レベルの妥当性を判断したようです。
状態での分子の IR スペクトルや N–H や O–H の結合長を計算したところ、 状態に比べて水素結合が強くなっていることが分かったそうです。そして著者らは、反応機構を次の 4 つに分類しました。2,6-DAI·(HO) で N -H···O から反応が進行する Type A、O
-H···O から反応が進行する Type A、O -H
-H ···N
···N から反応が進行する Type B、2,6-DAI·(HO) で N-H···O から反応が進行する Type C、O-H···N から反応が進行する Type D。
から反応が進行する Type B、2,6-DAI·(HO) で N-H···O から反応が進行する Type C、O-H···N から反応が進行する Type D。
結論としては、Type A のエネルギー障壁が 13.16 kcal/mol、Type C のエネルギー障壁が 13.14 kcal/mol で、クラスター中の水分子の数によらず、同様の反応機構で進行するそうです。
気になった点
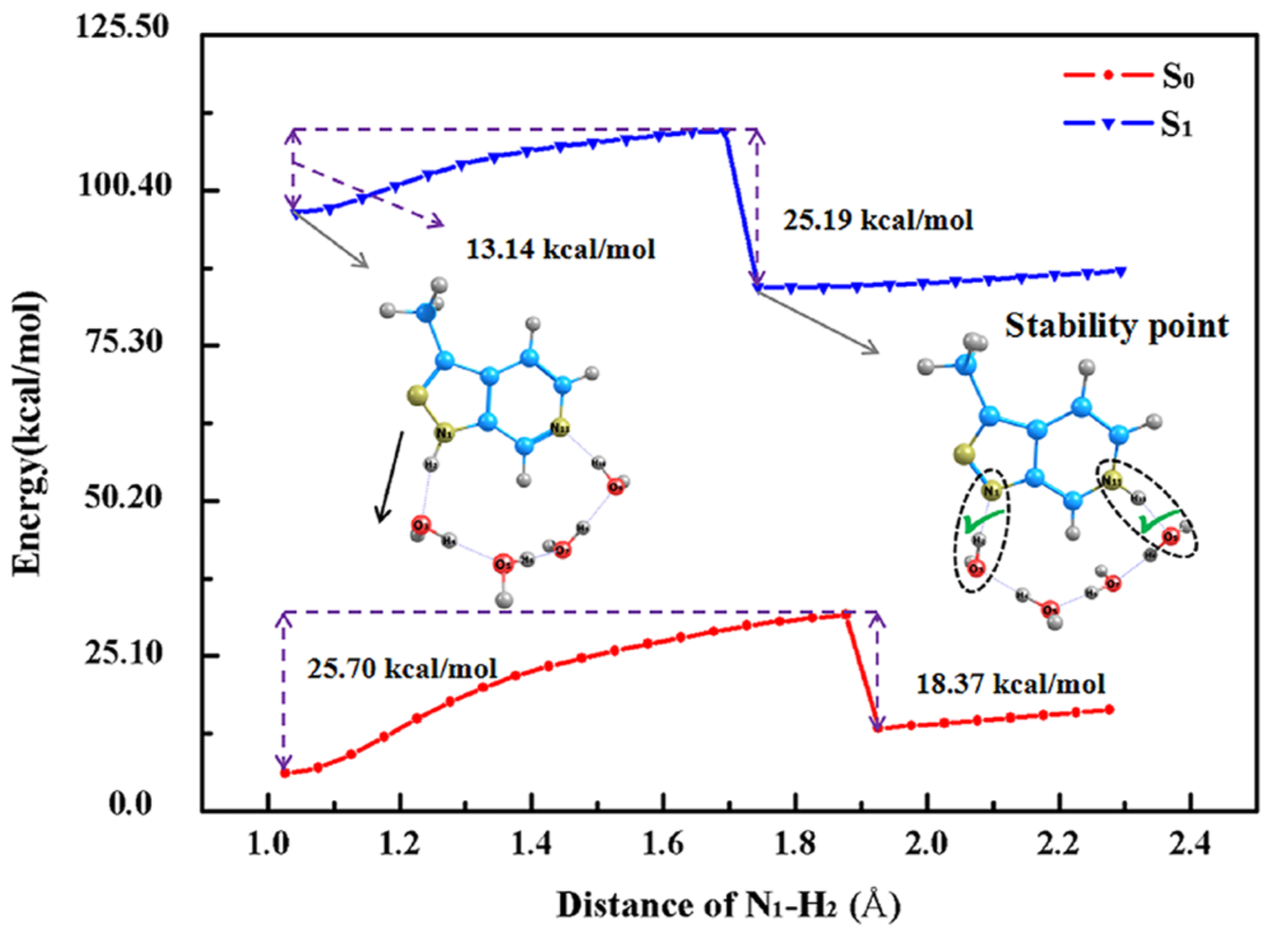
論文中の PES の図を転載
論文中で示されている PES の図ですが、どれも TS の直後にエネルギーがガクッと落ちています。こういう PES の時は、分子と水のコンフォメーションが急に変化していることが多く、遷移状態構造最適化を行おうとしても収束しない場合が多いです。N-H bond の scan ではなく、普通に IRC plot を見せれば良いのでは?と思ってしまいます。IRC の plot でも N–H bond の距離に応じたグラフが作れそうな気がします。
図中の 13.14 kcal/mol を活性化エネルギーとして Discussion に使用していますが、これは 遷移状態最適化後に IRC 計算によって得られた値なのかが気になります。こういう疑念を打ち消すためにも SI に座標を載せるべきだと思うのですが。。。
Type B についての Discussion もよく分かりません。活性化エネルギーが出ているのに、反応後の構造が収束しないと言っています。つまり、IRC 計算が終わっていないってことですよね?
他グループの JACS の論文 (参考文献 1 ) と違うことがわかったと言っているのに対して、計算の詳細部分に不明な点が多い論文だと思いました。
記事中に間違い等ある場合は、コメント欄、twitter またはメールにてお知らせいただけると幸いです。
参考文献
- “The Excited-State Triple Proton Transfer Reaction of 2,6-Diazaindoles
and 2,6-Diazatryptophan in Aqueous Solution”
Kun-You Chung, Yi-Han Chen, Yi-Ting Chen, Yen-Hao Hsu, Jiun-Yi Shen, Chi-Lin Chen, Yi-An Chen & Pi-Tai Chou
J. Am. Chem. Soc. 2017, 139, 6396−6402. DOI: 10.1021/jacs.7b01672
関連する記事
- Coenzyme B12 依存性酵素 ONIOM 計算の汎関数ベンチマーク
- Spinosyn A の生合成 〜[4 + 2] or [6 + 4] Cycloaddition?〜
- 遷移状態後の枝分かれでの Dynamic Effect
- ダブルハイブリッド密度汎関数理論 doubly hybrid density functional theory
- 正宗・Bergman 環化の計算化学【エンジイン】
- 【スパコン】Titan【アメリカ】
- gaussian scan の結果を解析するプログラム
- 原始地球上の非生物的条件下での核酸塩基の生成機構
- GRRM の振動計算の結果を Gauss View で解析する方法
- 【スパコン】天河二号【中国】
- GPU を用いた並列計算
- Rh(II) 触媒を用いたインドール合成の計算化学【反応機構解析】
- 構造最適化の閾値は、何を意味しているのか?
- 量子化学計算で a.u. を使う理由 〜Why Atomic Unit?〜
この場合、S1状態でのIRCを流すのは可能なのでしょうか