AutoDock は、タンパク質にリガンドをドッキングするドッキングシミュレーションに広く用いられています。他にもドッキングシミュレーション用のソフトはいくつもありますが、一番の特徴は無料であるということだと思います。
AutoDock は Windows でしか動かないと勘違いしている人をたまに見かけますが、Mac でも Linux でも動きます。BootCamp のような仮想化ソフトも必要ありません。
ネット上にいくつか AutoDock の使い方はありますが、少し古い部分もあるため、本記事で AutoDock Vina の使い方についてまとめることにしました。途中までは、pdf 形式の有名な AutoDock マニュアルと同じです。
無料で、かつ使い方も簡単な AutoDock ですが、そのドッキング結果は必ずしも正確ではありません。AutoDock で示された結果をあまり鵜呑みにしないほうが良いです。個人的には、実験の合間に片手間にできる簡易的なドッキングシミュレーションとしか捉えていません。
ドッキングシミュレーションを行う際には、ドッキングスコアの計算方法、対応している系をきちんと調べておく必要があります。
目次
AutoDock と AutoDock Vina の違い
AutoDock Vina がリリースされた当初は、AutoDock よりも高速・高精度なアルゴリズムである上に、マルチスレッドでの計算に対応しているということが売りにされてました(こちらのページ参照)。また、Input ファイルの作り方も Vina の方が簡単です。
しかし、その後 MPI/OpenMP を利用した CPU 並列化計算に対応した Multilevel Parallel Autodock4.2 や、GPU 並列計算に対応した AutoDock Software in Parallel with GPUs というものもリリースされました(管理人は、どちらも使ったことがありません。gpu 対応版については、 cuda 8.0 に対応していないため使うことができませんでした。一応インストール方法を本記事の一番下に書いておきました)。
必要なソフトウェア
ドッキングシミュレーションを行うのに AutoDock Vina を使います。ドッキングシミュレーション用の input file を作るのに MGL Tools というものを使います。結果の解析には MGL Tools または Pymol を使います。
mac でも linux でも使うことができます。管理人は windows を使ったことがないので、windows に関しては分かりません。
インストール方法
【Auto Dock Vina】
こちらのページよりダウンロードしてきた tar.gz ファイルを /usr に移動し、解凍します。
sudo mv autodock_vina_1_1_2_linux_x86 /usr sudo tar -zxvf autodock_vina_1_1_2_linux_x86
bin ディレクトリ内に vina という実行ファイルがあるので、/usr/autodock_vina_1_1_2_linux_x86/bin に path を通しておきます。
【MGL Tools】
次に、MGL Tools をこちらのページからダウンロードし、tar.gz ファイルを /usr に移動し、解凍します。
sudo mv mgltools_x86_64Linux2_1.5.6.tar.gz /usr sudo tar -zxvf mgltools_x86_64Linux2_1.5.6.tar.gz
bin ディレクトリ内に adt, pmv などの実行ファイルあるので、 /usr/mgltools_x86_64Linux2_1.5.6/bin に path を通しておきます。
【Pymol】
pymol はパッケージマネージャーで簡単にインストールできます。Fedora23 の場合には、
sudo dnf install pymol
AutoDock vina に必要なファイルの種類
計算を始めるにあたり以下の 4 つのファイルを作る必要があります。
- protein_rigid-part.pdbqt //タンパクの非可動部位のPDBQT ファイル
- protein_flex-part.pdbqt //タンパクの可動部位のPDBQT ファイル
- ligand.pdbqt //リガンドのPDBQT ファイル
- input.txt
input ファイルの拡張子は txt である必要はありません。
ドッキングシミュレーションでは、計算従事者が指定すべき項目がいくつかあり、それによって結果が左右されます。例えば、タンパク質内で特定のアミノ酸を可変部位といて指定し、残りの部分は固定して計算します。また、リガンドの自由度を自分で指定します。さらに、ドッキングシミュレーションで用いるタンパク質の構造が X 線結晶構造なのか?ホモロジーモデルなのか?MD で計算したものなのか?などでも結果が左右されます。
タンパクのPDBQTファイルの準備
まずは、ドッキングシミュレーションにて用いるタンパク質の pdb ファイルを用意します。Protein Data Bank から取得するのが一般的だと思います。
pdb ファイルを AutoDockTools (adt) で開きます。(Python molecule Viewer を開いてから AutoDock を選択するのではなく、直接 adt 開いたほうが早いです。)
まずは、color –> by atom type –> All geometry –> OK
{kind=link}
次に edit –> Hydrogen –> Add –> All Hydrogens
{kind=link}
データを保存 File –> save –> Write PDB –> OK
{kind=link}
次に、表示を消す。
{kind=link}
Grid –> Macromolecule –> Open から pdb ファイルを開く。
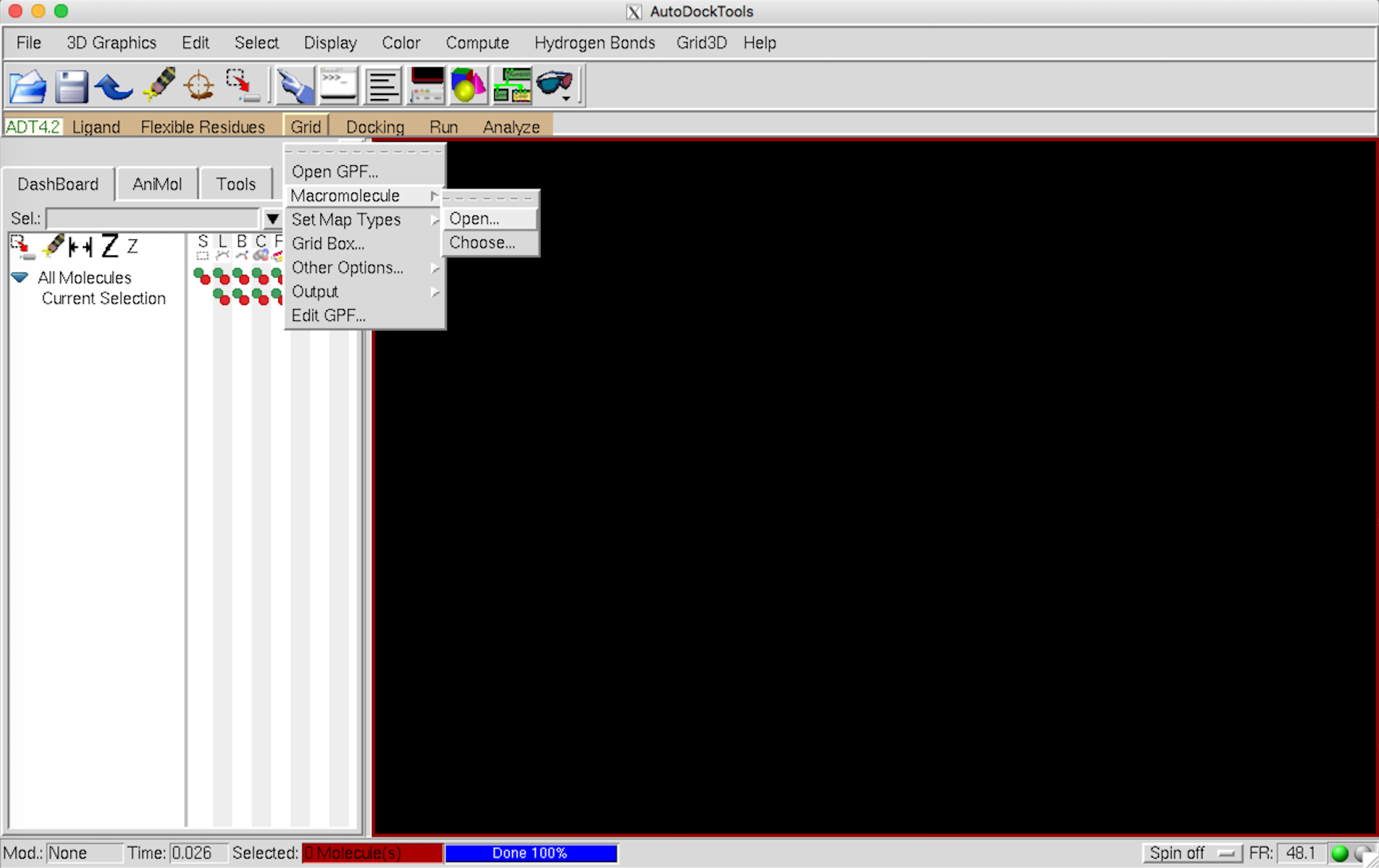{kind=link}
この際、いろいろと警告が出るが無視。自動的に pdbqt ファイルの保存へと進む。
再度表示を消す。
Flexible Residues –> Input –> Open Macromolecule で 先ほど保存したタンパクの pdbqt ファイルを開く。
左側の Select から動かすアミノ酸を選択していく。選択すると黄色くなる。MGL Tools はやたらと重たいので、選択するアミノ酸は Pymol などで事前に確認しておくことをお勧めします。
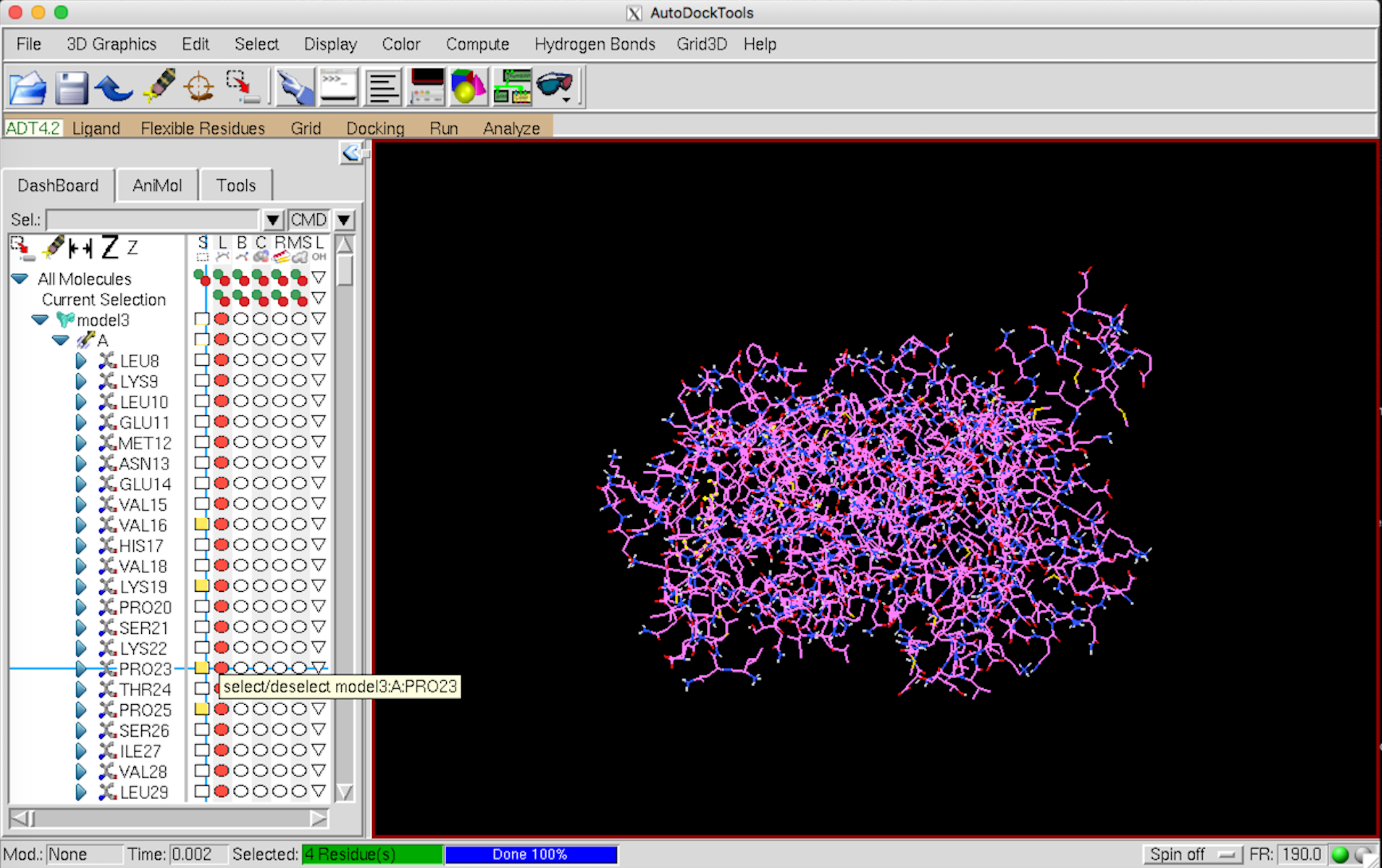{kind=link}
Flexible Residues –> Choose Torsions in Currently Selected Residues… を選択。
{kind=link}
Flexible Residues –> Output –> Save Flexible … で保存。これで protein_flex-part.pdbqt の準備は終了。
続けて Flexible Residues –> Output –> Save rigid … で保存。これで protein_rigid-part.pdbqt の準備は終了。
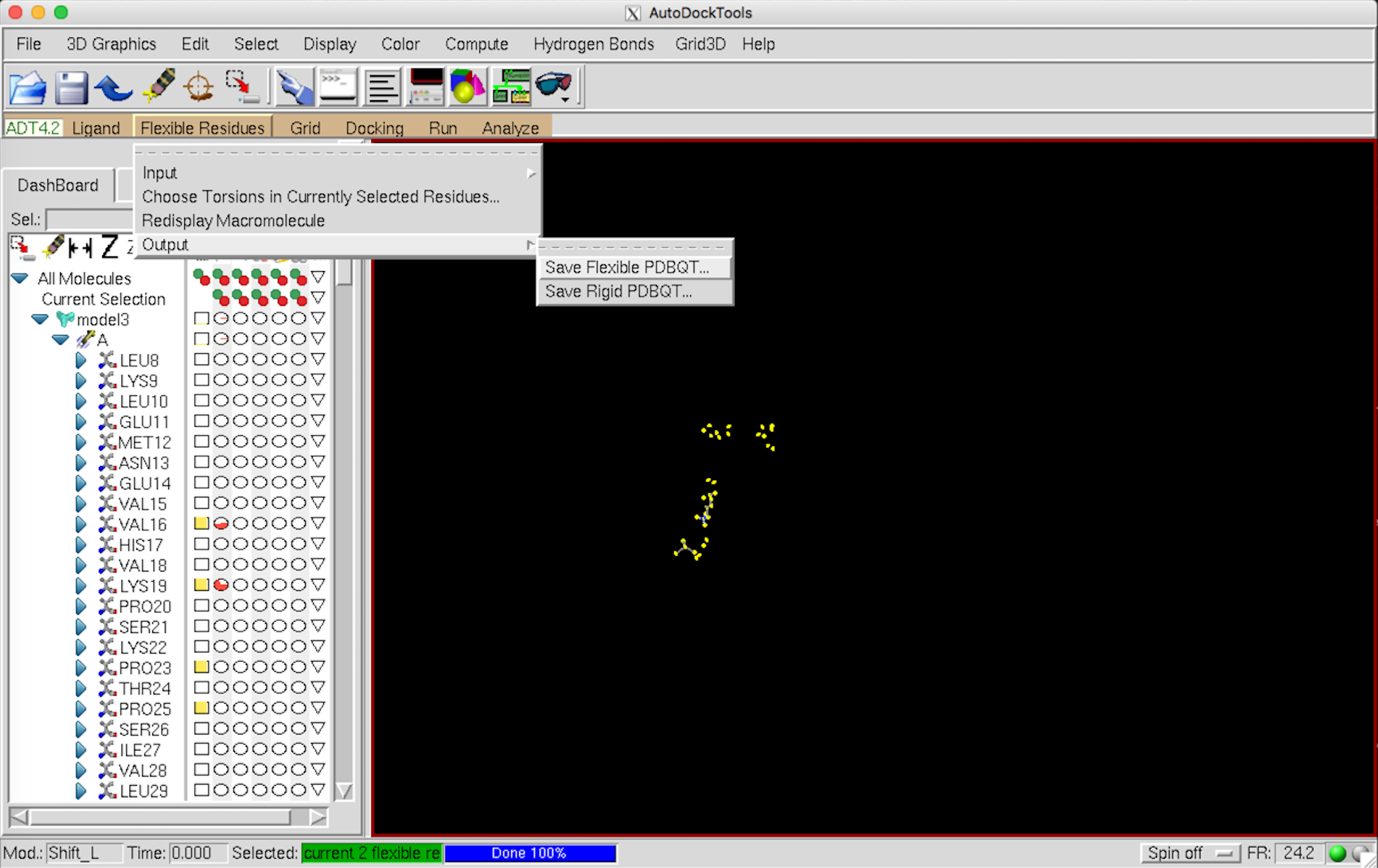{kind=link}
表示を消す。
リガンドのPDBQT ファイル
まず、gauss view などでリガンドを作成し、半経験的手法で構造最適化しておきます。
構造最適化後の座標を pdb ファイルで保存します。
次に Auto DockTools で Ligand –> Input–> Open でリガンドの pdb ファイルを開きます。
Ligand –> Torsion Tree –> Detect Root を選びます。
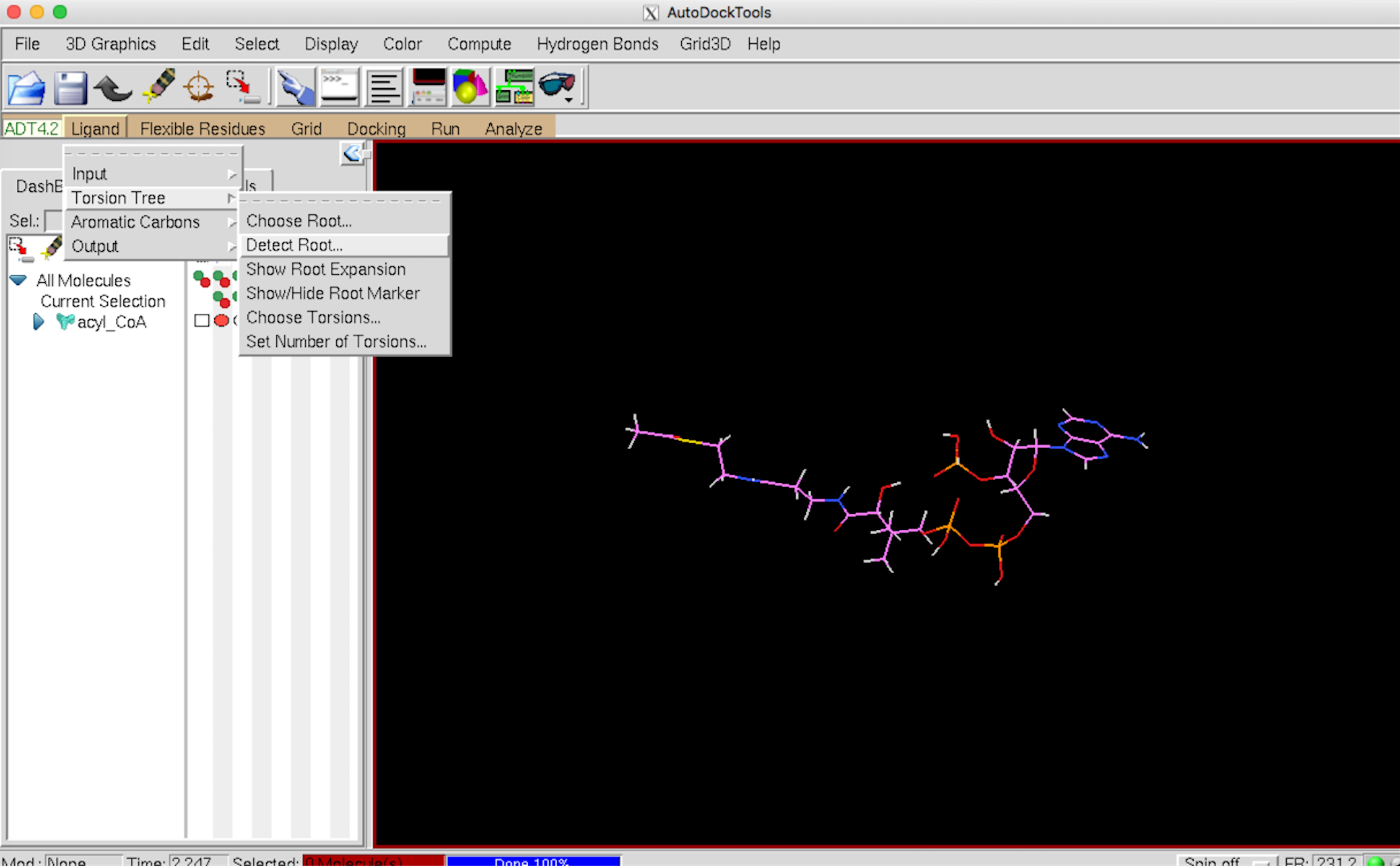{kind=link}
Ligand –> Output –> Save as PDBQT で保存します。これで ligand.pdbqt の準備は終了です。
表示を消します。
GRID BOX の設定
次にドッキングシミュレーションを行う探索範囲を指定していきます。無印の AutoDock と AutoDock Vina ではここから作業が変わります。
ここまでで作成してきたファイルを開きます。
Grid –> Macromolecule –> Open で protein_rigid-part.pdbqt を開きます。
Flexible Residues –> Input –>Open Macromolecule で protein_flex-part.pdbqt を開きます。
Ligand –> Input –> Open で Ligand.pdbqt を開きます。
Grid –> Grid Box を開きます。まずは、spacing を 1.000 Å に変更してください。
{kind=link}
結合部位に関する情報が明らかな場合は、Ligand.pdbqt と protein_flex-part.pdbqt のみ開いて、両方が Grid Box に入るように調整します。
結合部位に関する情報が何もない場合は、protein_rigid-part.pdbqt も表示させて、全てが Box 内に入るように調整します。
Grid Box が大きくても計算速度はそこまで遅くなりません。むしろ、Flexible Residues の残基数が増えたほうが計算時間は長くなります。
このとき設定した Grid Box の値をメモしておいてください。
INPUT ファイルの作成
以下に input ファイルの例を示します。これまで作成したファイルや Grid Box の情報を書き込みます。
receptor = protein_rigid-part.pdbqt flex = protein_flex-part.pdbqt ligand = ligand.pdbqt log = docking.log center_x = -0.759 center_y = -28.457 center_z = -7.032 size_x = 48 size_y = 70 size_z = 68 cpu = 8 exhaustiveness = 8 num_modes = 100 energy_range = 3
exhaustiveness, num_modes, energy_range などはいろいろと検討して調整してみてください。
無印の AutoDock では様々なファイルを作らなくてはいけなくて面倒ですが、Vina は非常にシンプルです。
AutoDock vina の 計算実行方法
計算を実行する場合は、以下のコマンドを使います。
vina --config input.txt
毎回 –config を実行するのは面倒なので、bashrc で alias の設定をおこうことをお勧めします。
計算実行中には termianl 上に進行状況が表示されますが、表示が不要な場合は
vina --config input.txt &
で実行してください。
結果の解析
pymol にて protein_rigid-part.pdbqt と ligand_out.pdbqt ファイルを開きます。
再生ボタンを押すと今回計算した全分子がコマ送りで表示されます。
{kind=link}
おまけ:GPU並列バージョンのインストール方法
こちらのページよりダウンロードしてきた tar.gz ファイルを /usr に移動し、解凍します。
sudo mv gpu*tar.gz /usr sudo tar -zxvf gpu*tar.gz
解凍されたファイルの中に DOCUMENT ディレクトリにインストール方法の解説書があるので、それを開きます。
less gpuautodock/document/INSTALL
以下、この説明書を読みながらインストール作業を進めていきます。
cd /usr/gpuautodock/src/autodock-4.0.1 sudo sh ./configure sudo make sudo make check sudo make clean
これが終わると、/usr/local/bin に autodock4 というバイナリーファイルが生成されています。
管理人は、ドッキングシミュレーションの専門家では無いため、記事中に間違いなどありましたら、コメント欄またはメールなどでご指摘いただければ幸いです。
View Comments (9)
薬理学の研究室で研究員をしているものです。
GPCRとリガンドのドッキングを、貴サイトを参考にしてうまくできました。ありがとうございました。
次の研究で、アプタマーと蛋白質のドッキングをしようとしているのですが、グリッド範囲を非常に広範囲に取らざるを得なくて、計算量が多いためか全くドッキングが進みません。
グリッド範囲を決める際のgridboxで表示させたときに、アプタマーの距離と蛋白質の距離が非常に遠くて広範囲に設定せざるを得ないのも一因かと思います。
もしよろしければ、この距離を詰める方法をご存じでしたらご教授いただければ幸いです。よろしくお願い申し上げます。
コメントありがとうございます。
アプタマーの pdb ファイルの座標を修正することで、アプタマーとタンパク質の距離が近くなりませんか?
分子構造は x, y, z 座標で指定されているので、全原子の座標に一律に数字を足したり引いたりして、タンパク質に近づくように平行移動させてやってください。
例えば、全ての原子の x 座標を +10 するなど。
座標の修正は、python などでスクリプトを書くと簡単に行えると思います。
そのあとで、グリッドボックスの設定を行ってみてください。
グリッドボックスが大きい場合には、計算に使う CPU のコア数を増やすことをお勧めします。
度々失礼致します。
R使いなので、rのbio3d というパッケージでPDBを読みこんで座標を編集するところまで来ました。
ここでもう一つお教え頂きたいのですが、ドッキングする二つの分子は重なるよう存在した状態でグリッドの範囲を指定しても良いのでしょうか。
中心をそろえれば、ドッキングの検証をするグリッドの範囲が最小限にできそうな気がするのですが。
ご教授頂けますと幸いです。
よろしくお願い申し上げます。
ご返信ありがとうございます。
二つの分子が重なった状態から AutoDock でドッキングしたことはありませんので、どのような結果が出るのか分かりません。
是非、ご自身で試してみて下さい。
早速お返事をありがとうございました。
なるほど、PDBの座標を修正すれば良いのですね。
ADTで何かできないかと思っていました。
スクリプト 調べてみて、良い例を見つけてみます。
グリッド数が、大きすぎるというアラートが出ていて全く進まず困っておりました。
skylake 8core のCPUで8コアフルに使っても全く進みませんでした。
なんとか座標をずらして、グリッド範囲の上限を超えないようにしてみます。
ありがとうございました。
医学系の大学院生です。
上記と同様に行ったのですが走らせても読み込むためのprotein_rigid-part.pdbqtを開けないというエラーばかり出るのですが、どうしたら改善できるのでしょうか。
今から使っていこうと思うのですが、なかなか進まず…
お教えください。。
工学系の大学生です。
inputファイルのexhaustiveness, num_modesはどういう意味なのでしょうか?マニュアルを見て調べようと思ったのですがどういう訳か現在つながらない状態です。
またこのサイトには載っていないですが他のオプションとしてseedがあると思いますがそちらの意味も教えていただければ幸いです。ただの乱数シードなのでしょうか?
医学系の大学院生です。
in silicoでの検討方法を勉強する中でたどり着きました。
こちらに記載されている内容を参考にドッキングをしてみていますが、ステータスバー(アスタリスク)が全く進まず、本当に動いているかどうか心配になっています。
CPUは、core i7 3770を用いています。
数日単位で、処理には時間がかかるんでしょうか。
お教えください。。
コメントありがとうございます。
並列処理であれば計算は数時間程度で完了します。flexible なアミノ酸残基の指定数や試行回数が多すぎると計算の進みが非常に遅くなります。