先日、読者の方から MD 計算を利用したコンフォメーション探索についてお問い合わせがありましたので、まとめてみました。
今回紹介する方法は私が考案したものではなく、香川大学の HP に書いてある方法とその補足になります。
今回の記事で使用する gromacs と antechamber のインストールは各自でお願いします。
目次
コンフォメーション探索はなぜ重要?
コンフォメーション探索と聞いて重要性のわからない人も多いかと思いますので、ここで少し詳しく説明したいと思います。一般的に、構造最適化をする際のポテンシャルエネルギー曲面は下図のように簡略化して表現されます。
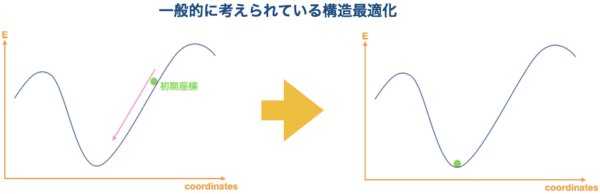{kind=link}
しかし、実際には下図のようにもっと複雑な形としています。見ていただいてお分りの通り、gaussian で構造最適化した結果、global minima ではなく local minima が得られてくる可能性は大いにあります。また、計算の結果得られてくる構造は初期座標に大きく依存してしまいます。
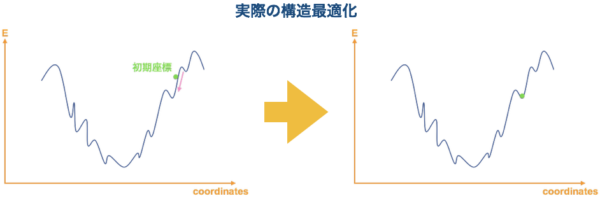{kind=link}
そこで、コンフォメーションを考慮した計算が必要となります。コンフォメーション探索ソフトは、分子内の結合角を色々と動かしてたくさんのコンフォマーを出してくれるソフトです。これをポテンシャル曲面上で考えると、多数の初期座標を与えてくれるということになります(下図左)。コンフォメーション探索のソフトで出された構造は、最終結果ではないことに注意してください!これらは初期座標なので、ここから更に QM 計算で構造最適化を行う必要があります。
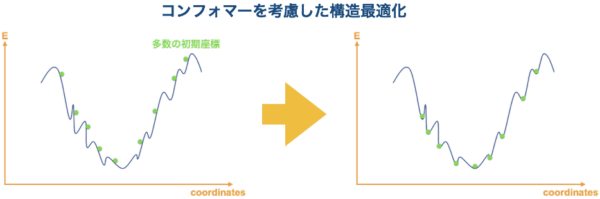{kind=link}
また、最安定なコンフォマーだけを用いて計算を進めていくというのも誤りです。これらのコンフォマーは互いに行き来できますので、存在確率の高いコンフォマー全てについて計算していく必要があります。
低分子化合物の計算を行う上で分子のコンフォメーション解析は欠かせません。適当に Gauss View 上で分子を作成して構造最適化するだけでは容易に誤った結果を導き出してしまいます(、、、というか、コンフォマーを考慮せずに遷移状態構造探索して結果出た、計算簡単とか言っている人は計算化学をなめ過ぎている気がします)。NMR 計算や光化学関連の計算、反応機構解析などなど全ての計算でコンフォマーを考えることは重要です。
頑張って計算しているのに計算結果と実験結果が一致しない。。。なーんて時はコンフォメーションが間違っている可能性も考えてみてください。
コンフォメーション探索のソフト
コンフォメーション探索のソフトは無数に出ています。
思いつく限りでも、Spartan, MacroModel, RDkit, Conflex, Tinker, Omega, Open Babel, Avogadro, Frog2 などがあります。
ちなみに、Gauss View 6 からコンフォメーション探索が可能になりましたが、gmmx という有料のプラグインを買わないと使用できません!Gauss View 自体高いのに、さらにお金取るの?って感じです。
管理人の個人的な感想としては、価格、使い勝手、計算結果ともに Spartan が良いと思います。ただし、Spartan にもいくつかのバージョンがあり、昔の Spartan はあまり良くありません。最新版が良いです。
次点で Conflex が良いと思いますが、欠点がいくつかあります。例えば、マクロ環をグニャグニャ動かしてくれるので、環上部を向いていた置換基が環の下向くようになってしまったりします。これは NMR 計算などでは重宝する機能だと思いますが、反応機構解析では邪魔です。また、カチオンやアニオンを認識することができなくて勝手に水素原子を足してくれたりします。。。あと聞いた話では、いくつかパラメータの入っていない金属などもあるそうです。。。確認していませんが。。。
MacroModel も良いですが、価格が、、、。
さて、コンフォメーション探索に興味を持ち、いざやってみよう!と思っても Spartan も Conflex も MacroModel も有料のソフトのため、すぐには取りかかることができません。また、Spartan、Conflex、MacroModel 以外のソフトは使い勝手が悪い上に、計算結果のコンフォマーも微妙なものが多いです。。。
無料かつ使いやすいソフトでコンフォメーション探索したいなーって人には、gromacs がおすすめです!
gromacs でのコンフォメーション探索の仕方
基本的な手法については、香川大学の HP を参照して下さい。このページに書かれている手順でコンフォメーション探索は十分行えますが、この操作を毎回繰り返すのは骨が折れます。そこで全部をまとめて実行するようにします。
以下のファイルを conformation_search.sh として保存してください。
#!/bin/sh #antechamber -fi mol2 -fo prepi -o ILV.prep -at gaff2 -rn ILV -c bcc -i $1 antechamber -i $1 -fi gesp -o ILV.prep -fo prepi -c resp -rn ILV -nc 1 rm ANTECHAMBER_* parmchk2 -i ILV.prep -o ILV.frcmod -f prepi -s gaff2 tleap -s -f tleap.in > tleap.out python ./ambergromacs.pygmx editconf -f gromacs.gro -bt cubic -d 1 -o box.grogmx grompp -f sa5cycles.mdp -c box.gro -p gromacs.top -o sa_test.tpr -maxwarn 1gmx mdrun -v -deffnm sa_testgmx grompp -f sa.mdp -c box.gro -p gromacs.top -o sa.tpr -maxwarn 1gmx mdrun -v -deffnm sa
上記のスクリプトでは、gaussian で電荷を計算したものを用いるように書いてあります。
分子の電荷の計算を antechamber に任せる場合は、”antechamber -fi mol2 -fo prepi -o ILV.prep -at gaff2 -rn ILV -c bcc -i $1″ という行を用いて “antechamber -i $1 -fi gesp -o ILV.prep -fo prepi -c resp -rn ILV -nc 1” をコメントアウトしてください。
一応書いておきますと、
vi conformation_search.sh
で上記の内容を書き込み保存した後、
chmod 755 conformation_search.sh
で実行権限を与えます。
実行する時は、
sh conformation_search.sh <file名>
です。
例えば、sample.esp というファイルを基に実行する時は
sh conformation_search.sh sample.esp
と実行してください。
また実行する際には、同じフォルダ内に ambergromacs.py と sa.mdp と sa5cycles.mdp と tleap.in の 4 つのファイルを置いておいて下さい。いずれも上述のウェブサイトにあります。
電子状態の計算
電子状態の情報が含まれている esp ファイルは、以下のインプットファイルで gaussian で計算します。ポイントとしては、iop の指定と pop=MK を使うこと、座標の下に一行空けて esp ファイル名を指定することです。10 分程度で計算は終わります。
%mem=16GB %nprocshared=16 # B3LYP/6-31+G** iop(6/41=10,6/42=6,6/50=1) pop=MK scrf=(iefpcm,solvent=ether) Molecule Name 1 1 C -4.64120000 0.18620000 0.00000000 H -5.26600000 0.26300000 -0.88980000 C -5.52780000 0.33230000 1.23210000 (中略) H -8.44040000 4.16790000 4.05910000 H -3.37830000 3.29310000 0.00000000 sample.esp
結合情報
mol2 ファイルの結合情報に起因するエラーが起きることがあります。
1.5 重結合は、全て単結合または二重結合に変えておきましょう!
計算結果の保存
gromacs で計算を実行すると多数のファイルが生成します。その中から sa.gro と sa.trr というファイルを見つけて下さい。この二つのファイルを drag&drop で PyMol2 で開いて下さい。PyMol1 ではなく、PyMol2 です。
PyMol2 上で Movie 形式でコンフォメーション探索の結果が見れると思います。
ここで、File > save molecule… を選択し、state のところで 0(all states) を選び保存します。保存形式は、sdf ファイルがおすすめです。
PyMol で保存した sdf ファイルを全て gaussian のインプットファイルに変換して、B3LYP/6-31+G(d,p) などで構造最適化、振動計算をします。その後、エネルギー値に基づいて結果を処理し、コンフォマーを絞り込んでいきます。
この操作で得られた コンフォマー達を用いて NMR 計算なり反応機構探索なりをしてください。
コンフォマーを素早く探索するソフトに関しては、以下の記事を参照して下さい。
gromacs を用いたコンフォメーション探索の妥当性
“コンフォメーション探索” と聞くと、簡単な計算と勘違いされる方も多いと思いますが、実は非常にチャレンジングな分野です。まだどの方法が良いかは結論が出ていませんし、様々な方法が報告されています。記事内で紹介したほとんどのソフトは MMFF を用いていますが、実は PM7 の方が良いよ!なんて論文もあります。
“A sobering assessment of small-molecule force field methods for low energy conformer predictions” Ilana Y. Kanal, John A. Keith, Geoffrey R. Hutchison, Int. J. Quant. Chem. 2018, 118, e25512. DOI: 10.1002/qua.25512
また、QM でのコンフォメーション探索の研究も行われています。
参考: QM でのコンフォメーション探索
gromacs を用いたコンフォメーション探索が妥当かどうかは分りませんが、spartan や conflex などに比べると劣ることだけは確かです。しかし、初期座標をたくさん得ると言う観点から言えば、十分です。
関連する記事
- ニューラルネットワークを利用した粗視化シミュレーション コンフォメーション探索
- 構造最適化の閾値は、何を意味しているのか?
- QM でのコンフォメーション探索
- ダブルハイブリッド密度汎関数理論 doubly hybrid density al theory
- B97 & B97 系汎関数【ωB97X-D など】
- DFT は、正確な関数を目指す道から外れつつある
- 機械学習を利用した IR スペクトルの解析
- アクセル・ベッケ Axel D. Becke
- Rh/Ru を用いた σ-bond activation に適した汎関数!
- NHC-Pd 触媒を用いた溝呂木・ヘック反応に最適な汎関数!
- マーティン・ヘッドゴードン Martin Head-Gordon
View Comments (0)