計算化学を用いた反応機構解析を行う上で、コンフォメーション探索は非常に重要です。これまでも本サイトではコンフォメーション探索についての記事を公開してきました。
一般的にコンフォメーション探索は、以下のステップで進めます。
- Gauss View などの molecular viewer で構造式を書く。
- PM7 などで簡単に構造最適化する。
- MM でコンフォマーを大量に発生させる(Spartan, Conflex, Macromodel などを用いるのが一般的。Gromacs でもコンフォマーを発生させることが出来る)。
- 全てのコンフォマーを量子化学計算で構造最適化する。
- エネルギー順に並び直し、最安定なものから X kcal/mol を選抜し、同一コンフォマーを取り除く。
今回の記事では、上記 5 の手順を効率化するプログラムについてご紹介します。
後処理の問題点
- コンフォメーション探索後の log ファイルは、100 個前後ありますので、手作業でこれらを処理することは非常に面倒です。
- MM では違う構造であっても、DFT で構造最適化すると同じ構造になってしまうものも多数含まれています。これらを molecular viewer で一つ一つ開いて同一コンフォマーかどうかを判定するのは非常に面倒です。
今回、上記 2 つの問題点を解決するプログラムを書きました。実装した機能は以下の二つです。
- エネルギーが低い順に log ファイルを並べ換える
- 同一コンフォマーかどうかを判定する
ソースコード
ソースコードは、下のダウンロードボタンをクリックしてダウンロードして下さい。以下のファイルが含まれています。
- conf_header.h
- conf_main.cpp
- conf_judge.cpp
- conf_other_function.cpp
- conf_overlaying.cpp
- conf_reader.cpp
- Makefile
コンパイルするときは、terminal で make と打ってください。conf-overlay という実行ファイルが生成します。
実際に使用している様子
実際に 102 個の log ファイルを処理している様子をご覧ください。これらの log ファイルは、gromacs で生成したコンフォマーを DFT 計算で opt & freq したものです。
2 秒程度で、全てのコンフォマーの重ね合わせ計算を行い、同一コンフォメーカどうかを判別しています。
なお、上の動画では実行時間が 2 秒程度となっておりますが、コンパイルの仕方でだいぶ速度は変わります。オプション指定なしでの g++ でのコンパイルだと実行時間は 20 秒程度かかってしまうのでご注意を!
結果の見方
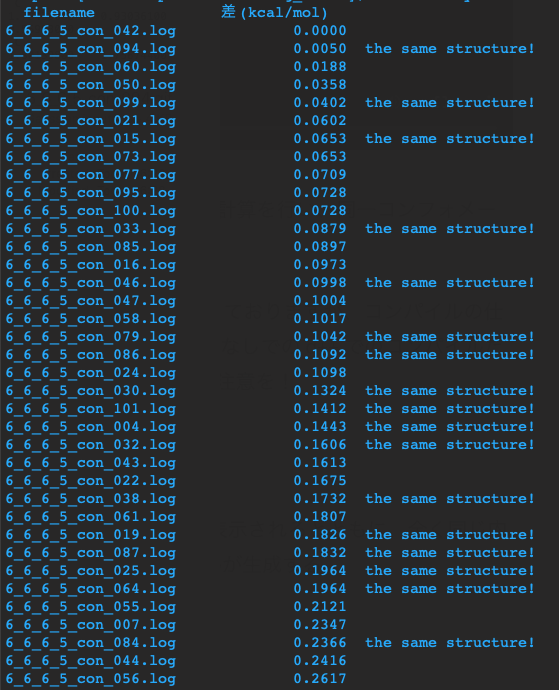
プログラムを実行すると terminal 上に結果が表示されるとともに、全く同じ内容の記述された energy.txt という出力ファイルが生成します。
下図のように、左のカラムにファイル名、次のカラムには最安定なコンフォマーとのエネルギー差(kcal/mol)、そして上行のコンフォマーと同一構造である場合は “the same structure” と表示されます。
下の例の一行目と二行目では、6_6_6_5_con_042.log と 6_6_6_5_con_094.log のエネルギー差は 0.0050 kcal/mol で同一コンフォマーであると表示されています。
アルゴリズムについて
フォルダ内の log ファイル名を読み込んでエネルギーの安定なもの順に並べ換える部分については、簡単なので説明を省略します。
以下、分子の重ね合わせについてのアルゴリズムについてです。
全くの同一コンフォマーであっても、gaussian の計算中に分子が回転してしまうことがあるため、3 次元座標内での位置は異なります。例えば、下図の二つのコンフォマーは全くの同一構造ですが、回転してしまっており、一目で同じコンフォマーかどうかを判別するのは困難です。

同一コンフォマーの判別には、分子の重ね合わせが有効です。このような二つの分子を重ね合わせる際には、以下の 5 つのプロセスが必要です。
- 分子内から 3 つの原子 A, B, C を選ぶ
- 原子Aが原点に来るように分子を平行移動
- 原子Bが XY 平面上に来るように、分子全体を X 軸周りに回転させる
- 原子Bが X 軸上に来るように、分子全体を Z 軸周りに回転させる
- 原子Cが XZ 平面上に来るように、分全体子を Z 軸周りに回転させる
上述の操作を行うと原子 A, B, C で構成される三角形はXZ 平面上にあり、かつ原子 A は原点、辺 AB は X 軸上にあります。
この操作は、「もし同一コンフォマーならば、3 つの原子が重なっていれば、同様の平行移動・回転移動の操作を施した他の原子も当然ぴったり重なるはずだ」という仮定に基づいています。
このアルゴリズムは、conf-overlaying.cpp というファイル名に書いてあります。
また、重ね合わせ操作の後に、二つのコンフォマー間で分子骨格を構成する炭素の RMSD を計算します。今回のプログラムでは、「エネルギー差が 1 kcal/mol 以下かつ RMSD が 1 原子あたり 0.05 Å 以下の場合、同一コンフォマーである」という閾値を設けています。
さいごに
何か不具合のある場合は、コメント欄によろしくお願いいたします。
spartanで発生させたコンフォマーをgaussianでopt freqした後、
Ubuntu(wsl) 上でmakeでコンパイルして稼働させましたが、
terminate called after throwing an instance of ‘std::bad_alloc’
というエラーを吐いてしまいます (それも12時間後に)
macでも試しましたが、メモリを食い尽くして止まったままでした。
どうすればよろしいでしょうか